







qp.qchem.meanfield¶
- meanfield(symbols, coordinates, name='molecule', charge=0, mult=1, basis='sto-3g', package='pyscf', outpath='.')[source]¶
Generates a file from which the mean field electronic structure of the molecule can be retrieved.
This function uses OpenFermion-PySCF plugins to perform the Hartree-Fock (HF) calculation for the polyatomic system using the quantum chemistry packages
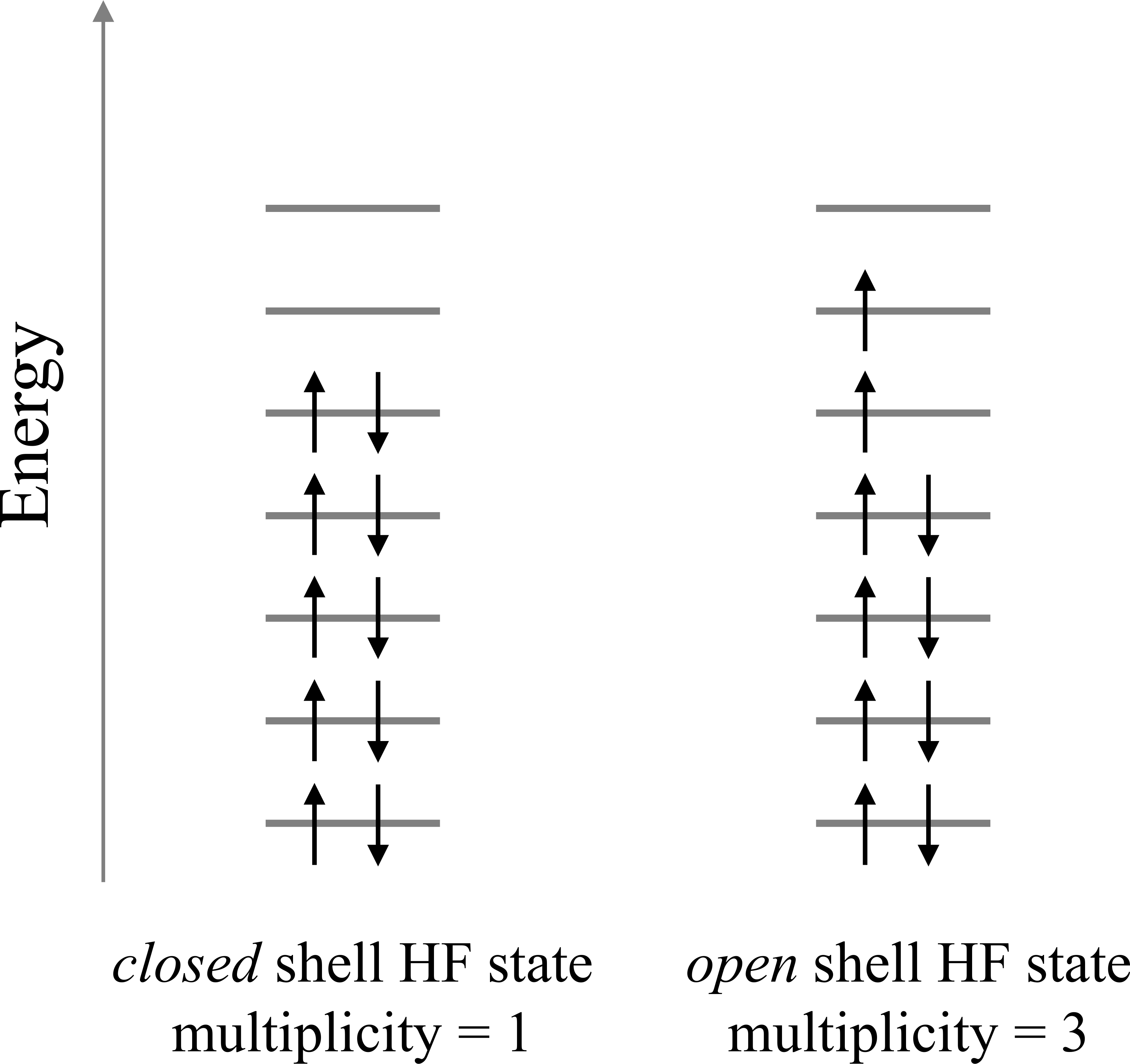
PySCF. The mean field electronic structure is saved in an hdf5-formatted file.The charge of the molecule can be given to simulate cationic/anionic systems. Also, the spin multiplicity can be input to determine the number of unpaired electrons occupying the HF orbitals as illustrated in the figure below.
- Parameters:
symbols (list[str]) – symbols of the atomic species in the molecule
coordinates (array[float]) – 1D array with the atomic positions in Cartesian coordinates. The coordinates must be given in atomic units and the size of the array should be
3*NwhereNis the number of atoms.name (str) – molecule label
charge (int) – net charge of the system
mult (int) – Spin multiplicity \(\mathrm{mult}=N_\mathrm{unpaired} + 1\) for \(N_\mathrm{unpaired}\) unpaired electrons occupying the HF orbitals. Possible values for
multare \(1, 2, 3, \ldots\). If not specified, a closed-shell HF state is assumed.basis (str) – Atomic basis set used to represent the HF orbitals. Basis set availability per element can be found here
package (str) – Quantum chemistry package used to solve the Hartree-Fock equations.
outpath (str) – path to output directory
- Returns:
absolute path to the file containing the mean field electronic structure
- Return type:
str
Example
>>> symbols, coordinates = (['H', 'H'], np.array([0., 0., -0.66140414, 0., 0., 0.66140414])) >>> meanfield(symbols, coordinates, name="h2") ./h2_pyscf_sto-3g